

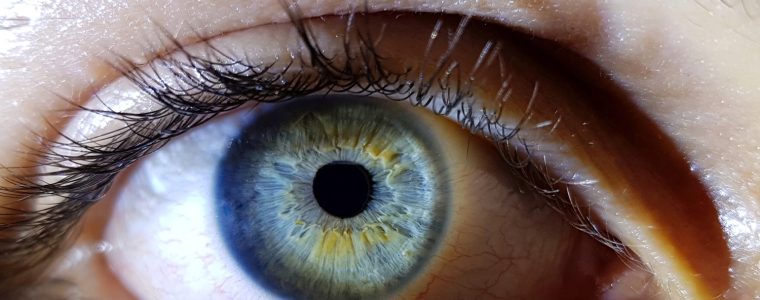

Choroideremia is a rare genetic disease linked to the X chromosomecharacterised by chorioretinal dystrophy and determined by sequence variations or deletions in the CHM gene. This gene codes for the protein REP-1 (Rab escort protein) essential for post-translational activation and intracellular localisation of Rab GTP-binding proteins that control vesicular trafficking in secretory and endocytic pathways. REP1 activity in human choroideremia appears to affect only the retinal pigment epithelium (RPE) of the eye, leading to progressive degeneration of the choroid, RPE and retina. The estimated prevalence of the disease is 1 in 50,000/100,000 individuals, with a male predominance. However, it is likely that the pathology is not correctly diagnosed because of its similarities to other eye disorders. It is believed that choroideremia accounts for about the 4% of all causes of blindness.
The first symptom of this condition is usually impairment of the night visionwhich may occur in early childhood. This is followed by the progressive narrowing of the field of view (tunnel vision), as well as a decrease in the ability to see details (visual acuity). Visual impairment in choroideremia worsens over time, but the progression occurs differently depending on the individual, however, all affected individuals will reach blindness in later life.
Choroideremia study published in Genes
A very recent review published in Genes offers an interesting overview of the new knowledge about clinical phenotyping and molecular genetic testing for choroideremia, discussing alternative molecular therapies, including the possibility of CRISP gene editing.
The molecular mechanisms underlying choroideremia are well known: the absence or reduction of prenylation in REP1 activity disrupts normal intracellular trafficking pathways leading to the accumulation of toxic products, premature retinal degeneration and reduced visual acuity. It is logical to assume, therefore, that replacement by gene therapy of REP1 in retinal tissue could restore cell function and slow down the degeneration of the disease. Several clinical trials using the adeno-associated viral vector AAV are currently underway to test this hypothesis. Molecular replacement therapy involves the sub-retinal delivery of AAV2-REP1 to the surviving central islands of the retina, this approach has shown promising safety and efficacy results, so that some of these trials have even reached phase III.
The review also addresses the complex interactions that occur between different retinal cell types during the pathogenesis of choroideremia and for which it is difficult to establish the exact order in which they lead to the degeneration of RPE, photoreceptors and choroid. It seems likely that the RPE is directly influenced by the loss of REP1 and is a key factor in pathogenesis of the disease, but the importance of primary or secondary degeneration of photoreceptors is still unclear. A better understanding of these mechanisms would be important in order to understand what triggers the onset of clinically significant degeneration and how the rate of degeneration in each cell type is affected following treatment.
Source:
JC Kapetanovic et al. Molecular Therapies for Choroideremia. Genes 2019, 10, 738.
Dr. Carmelo Chines
Direttore responsabile