


Age-related macular degeneration (AMD) is the leading cause of severely reduced visual acuity in people over 60 in the western world. It is a complex disease that recognises several risk factors such as age, gender, race, light exposure, diet, cigarette smoking and vascular disease.
Recently, other elements seem to play an important role in the pathogenesis of the disease: variations in DNA sequences coding for complement factors, modifications of TMP-3 and activation of inflammatory cytokines. Knowledge of new factors involved in the pathogenesis of this disease may in the future pave the way for new therapeutic strategies to combat AMD.
INTRODUCTION
Age-related macular degeneration (AMD) is a degenerative disease involving the macular region in people over the age of 55. Its severity and frequency make it the leading cause of severe reduction in visual acuity, termed 'legal blindness', in people over 60 in the Western world. AMD can be divided clinically into a dry form (dry), the most frequent and least severe, and an exudative form (wet). The latter accounts for approximately 20% of AMD, but is the cause of approximately 90% of the severe cases of irreversible reduction in visual acuity that are always linked to this disease. This disease involves a progressive loss of the central portion of the visual field, with sparing of the peripheral one, which means no longer being able to read, drive, recognise people's faces, bus numbers, etc.
According to fairly recent estimates, about 500,000 new cases of exudative AMD appear each year worldwide and this number, with the increase in average life expectancy, will tend to rise dramatically in the coming decades. It is estimated that 8.5 million Americans currently suffer from the disease, and by 2020, some 7.5 million new cases are expected in the United States.
Age-related macular degeneration is a complex disease to study because of the potential involvement of various factors including demographic, environmental, age-related risks, gender, race, light exposure, diet, smoking and cardiovascular disease.
Macular degeneration and/or macular dystrophies can be classified into juvenile and senile forms. Regarding the molecular genetics of juvenile macular dystrophies, research over the past 10 years has contributed significantly to the understanding of these forms. Clinical features and the genes involved have been identified. Much of the progress in understanding juvenile macular dystrophies is related to the early onset of the disease. Collectively, these studies have led to the identification of the numerous genes involved in the visual process.
It was recently discovered that single nucleotide polymorphisms and DNA sequence variations, found within the complement factor H (CFH) gene, are strongly associated with the development of AMD in Caucasians. The same authors found the involvement of other genes in AMD, including the VEGF gene and two genes involved in lipid metabolism. A single nucleotide polymorphism of CFH, Tyr402His, was associated with approximately 50% of cases of AMD. Another gene involved in the complement cascade has been associated with the pathogenesis of AMD. This gene, located on chromosome 6, produces a protein called Factor B, which activates complement. A third gene involved (LOC387715) has been mapped on chromosome 10; the risk of developing AMD appears to be related to the variation of a single amino acid in the encoded protein, which is unrelated to the complement cascade. Genetic analyses indicate that the effect of LOC387715 is independent of that of factor H, but equally important, possibly contributing to almost 40% of cases of AMD. Pericak-Vance et al. showed a relationship between the high-risk variant of LOC387715 and cigarette smoking, a major environmental risk factor for AMD. The data reported so far show how important it is that scientific research can, in terms of prevention, develop a new therapy capable of halting the dramatic development of this truly disabling disease.
As far as dry AWD is concerned, several studies exist:
- The Age-related Eye Disease Study (AREDS):
US multicentre trial set up to evaluate whether vitamins, antioxidants and/or minerals reduce the development and/or progression of the disease.
- The Prophylactic Treatment of Age-related Macular Degeneration (PTAMPD) Clinical Trial:
Randomised pilot study on the effectiveness of laser treatment of soft drusen at the posterior pole, which demonstrated, at two years, a reduction in drusen in the 70% of treated subjects and, consequently, an improvement in visual acuity. For exudative AMD, new proposals have been added to the traditional laser treatment based on the destruction of neovascular areas, resulting in anatomical and functional damage to the treated retina:
- Focal treatment of feeder vessels: based on low-intensity photocoagulation with argon-laser and/or diode laser (infrared) of afferent neovases (feeder vessels) of neovascular membranes, identified by dynamic angiography with sodium fluorescein and indocyanine green with laser-scanning ophthalmoscopes.
- Transpupillary Thermal Therapy (TTT): is mainly indicated for 'occult' forms, it exploits the thermal effect of the diode laser to cause occlusion of the neovascular membranes without destroying the overlying retina. It is however a technique that still needs further study and modification; the difficulty of this technique is linked to the lack of data on the correct dose to use. It has been introduced for the treatment of small melanomas of the posterior pole choroid.
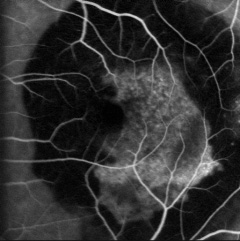
- Surgical removal: the results are rather disappointing in AMD due to iatrogenic damage on the retinal pigment epithelium. Better results are obtained with retinal 'rotation' (Machemer, Erkhardt and Todt) and 'limited translocation' (De Juan, Tano), which displaces the foveal region from the underlying membrane so that the latter can be photocoagulated.
- Radiation therapy: aims to prevent the proliferation of endothelial cells in choroidal neovases and induce their closure by ionising radiation. However, randomised clinical trials (DAR) have not yielded good results.
- Photodynamic therapy (PDT): Before its advent, there were few intervention options for subfoveal choroidal neovascularisations. Three multicentre trials, TAP, VIP and VIT (the latter carried out in Italy and coordinated by the Ophthalmology Clinic of the San Paolo Hospital in Milan) have demonstrated its efficacy.
- Drug therapy: these are corticosteroids (Triamcinolone Acetonide) and drugs with anti-angiogenic and angiostatic action such as the anti-VEGFs AVASTIN, MACUGEN and LUCENTIS, all administered intravitreally.
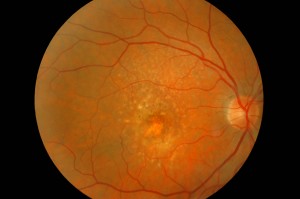
CLINICAL FEATURES
AMD, as already mentioned, has been classified into two clinical conditions: a dry form and a wet form. Choroidal neovascularisation is characteristic of the wet form, a stage found in approximately 10% of cases. Although both forms of AMD can cause visual loss, the wet form is involved in approximately 90% of cases of severe visual loss. It is also generally accepted that the wet form of AMD is preceded and develops from the dry form. In contrast, the dry form is much more common and is characterised clinically by the presence of macular drusen, which are localised deposits between the EPR and Bruch's membrane, and by geographic atrophy, characterised by cell death of the EPR with atrophy of the overlying photoreceptors.
AMD is usually not clinically evident before the age of 50. From autopsy findings, AMD is found in 33% of the eyes of persons over 65 years of age and in approximately 30% of persons over 75 years of age. The prevalence of the disease ranges from 1.6% in individuals aged 52 to 64 years to 27.9% in those over 75 years. The prevalence of AMD is negligible at age 50 and reaches 6% at age 80.
These discrepancies can be explained, in part, by the fact that the eye, like other organs, undergoes a normal ageing process independent of AMD, which, unlike juvenile macular dystrophies, can make diagnosis more difficult. In addition, AMD is associated with cardiovascular disease and other major disorders, so it is possible that many potentially affected individuals may die of cardiovascular disease before AMD manifests itself, thus lowering the prevalence of the disease in 80-year-olds.
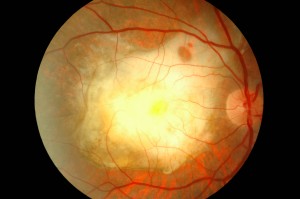
DEMOGRAPHIC AND ENVIRONMENTAL RISK FACTORS FOR DMLE
Age, gender, race, light exposure, concomitant cardiovascular disease, diet, and smoking are possible risk factors for AMD. AMD has a higher prevalence in the white population than in the non-white population. There are conflicting studies on the effect of chronic exposure to light (ultraviolet or visible) on the retina.
DRUSEN AS IMMUNOLOGICAL MARKERS OF DMLE
The molecular and cellular constituents of drusen have been extensively analysed. Much of the material found in drusen is synthesised by cells normally found in the eye, but some of the material derives from extraocular sources. For example, complement, lipids, lipoproteins B and E are common constituents of ocular drusen and atherosclerotic plaques, suggesting that the same biochemical and immunological processes may be involved in both conditions. For example, amyloid beta, a major inflammatory component of plaques in Alzheimer's, is also found in drusen. Hageman et al. proposed that drusen may be the product of a localised inflammatory response following EPR damage involving HLA antigens and the complement system. The hypothesis is based on the observation of drusen in MPGNII, a renal disease in which complement-mediated immunological dysfunction leads to renal failure. In this condition, the drusen are identical to those in AMD.
Drusen are focal deposits of extracellular material, located between the basement membrane of the EPR and the collagen layer of Bruch's membrane. Drusen are important risk factors and biological indicators of age-related macular degeneration.
They are commonly observed in individuals over 60 years of age and in the clinical context of AMD. The size, number, extent and confluence of drusen are important determinants in the risk of developing AMD (Pauleikhoff et al., 1990). The presence of soft, large and/or confluent drusen correlates with the occurrence of choroidal neovascularisation, with a relative risk of 2.1 in eyes with five or more drusen and 1.5 in eyes with one or more large drusen (Macular Photocoagulation Study Group, 1993).
The presence of drusen is associated with various visual deficits that occur prior to visual acuity loss: changes in colour and contrast sensitivity, macular functional recovery, central visual sensitivity, and spatiotemporal contrast sensitivity (Frennesson et al., 1995; Holz et al., 1995; Midena et al., 1997, 1994; Stangos et al., 1995; Tolentino et al., 1994).
Several studies have been undertaken with the aim of determining the composition of drusen, in the belief that understanding the constitution of a disease-related deposit could provide new information about the pathogenetic process itself.
Lipids
Donders first observed the presence of lipids in drusen in 1854. Using histochemical and enzymatic techniques, it was concluded that the lipid component of drusen probably comprised glycolipid molecules, cerebrosides and/or gangliosides (Wolter and Falls, 1962; Farkas et al., 1971; Pauleikhoff et al., 1992). Pauleikhoff et al. (1992) suggested the existence of distinct classes of drusen, some with more 'hydrophilic' characteristics, others more 'hydrophobic'. Holz et al. (1994) found a higher lipid concentration in the macular region than in the peripheral regions of Bruch's membrane; the characteristics of these lipids would suggest their cellular rather than vascular origin. In contrast, Curcio et al. (2001) describe the presence of both esterified and non-esterified forms of cholesterol in Bruch's membrane and drusen: these authors hypothesise that the considerable amount of esterified cholesterol in drusen may imply a vascular (plasma) source of the lipids associated with drusen and Bruch's membrane. Studies by Haimovici et al. (2001) prove the presence of cholesterol esters in drusen.
Despite numerous studies, the source (cellular, vascular or combined) of drusen-associated lipids remains an enigma. However, Hageman et al. ascertained that the genes for some drusen-associated lipoproteins (such as lipoprotein E) are transcribed locally, whereas other molecules (such as the P-component of amyloid) seem to have a hepatic origin, and would reach Bruch's membrane via the circulation. Drusen-associated lipids could similarly have a heterogeneous origin.
Carbohydrates
Farkas et al. first admitted the existence of glycolipids within drusen (1971). Kliffen et al. (1994; 1996), Farkas et al. (1971; 1996) identified glycoconjugates rich in glycosaminoglycans in deposits within Bruch's membrane.
Mullins et al. (1997) identified glycocompounds with endings consisting of glucose/mannose, N-acetylglucosamine, sialic acid as the carbohydrates most commonly represented in drusen, both soft and hard.
When the sialic acid residues of the carbohydrate endings associated with drusen were removed by neuraminidase, galactosamine disaccharides were found (Mullins and Hageman, 1999). These disaccharides were confined to a central core consisting of glycoproteins with carbohydrates bound via O-glycosidic bonds. The presence of these core domains within drusen is indicative of a role of the core in the earliest stages of drusen development, similar to the growth of a pearl in an oyster shell.
Protein
The protein constituents of drusen first identified by immunohistochemical methods included ubiquitin (Loeffler and Mangini, 1997), integrins (Brem et al., 1994), tissue inhibitors of metalloproteases (Fariss et al, 1997), glycosylation end products (Ishibashi et al., 1998), beta-amyloid (Loeffler et al., 1995), fibronectin (Pauleikhoff et al., 1992) and C1q (van der Schaft et al., 1993). In recent years, vitronectin (Hageman et al., 1999), the P-component of amyloid, apolipoprotein E, factor X, lambda chains of immunoglobulins, complement activation factors such as the C5b-9 complex, and MHC class II antigens have been identified (Johnson et al., 2000; Mullins et al., 2000; Mullins and Hageman, 1997). Molecular heterogeneity does not correspond to ultrastructurally or clinically defined phenotypes (Hageman and Mullins, 1999).
Virtually all recently discovered proteins in drusen are in some way associated with inflammation and/or other immune-associated processes. Some are classic acute phase proteins, while others are components of the complement cascade, or inhibitors of the complement membrane attack pathway. Still others are associated with immunological activation, coagulation, and fibrinolysis. Moreover, many of these molecules are common in pathological deposits associated with other diseases such as Alzheimer's disease, atherosclerosis, elastosis, amyloidosis, and glomerulonephritis (Mullins et al., 2000), so it was thought possible that common pathogenetic pathways might be involved in their formation.
Cellular Components
According to traditional models of drusen formation, any cellular material within them was of EPR origin. In fact, cell fragments and organelles derived from the EPR, even whole cells, can be found in the 'initial' drusen. In addition, EPR vesicles have been described that extend across the EPR basement membrane within drusen or at the sites of their subsequent formation (Ishibashi et al., 1986). Constituents of the EPR, such as lipofuscin and melanin, can sometimes be observed in small, early drusen.
A new and potentially very significant observation is that molecules normally associated with cells, including HLA-DR and CD-specific antigens, associate with drusen. These molecules are often located in solitary, core-like domains in the context of drusen (Mullins et al., 2000). Recent immunophenotypic analyses have documented that these cores derive from cellular extensions of choroidal dendritic cells, powerful antigen presenting cells associated with various immunomodulation processes. Moreover, these dendritic cell processes are typically associated with the EPR vesicles already described. These data suggest for the first time that the drusen formation process may be cell-mediated and that specific immune-mediated pathways may play significant roles in these events.
mRNA
Many of the molecules associated with drusen, recently identified using immunohistochemical methods, are mainly synthesised in the liver. Their escape from the capillaries of the choroid and subsequent aggregation along Bruch's membrane is a potential initiation towards their deposition within the drusen. However, certain local cell types in the neuroretina, EPR and/or choroid, which are located close to Bruch's membrane, may also have the ability to synthesise part of these molecules. Evidence for the gene transcription of some drusen-associated molecules in the neuroretina, EPR and choroid has recently been obtained with the use of PCR techniques: several PCR products obtained from drusen-associated mRNAs (TIMP-3, apolipoprotein E, vitronectin, C3, C5, C9) were found in the neuroretina, EPR, choroid and/or isolated human EPR cells (Alexander et al, 1990; Anderson et al., 2001; Hageman et al., 1999; Mullins et al., 2000). Quantitative studies have also been undertaken to identify the expression levels of drusen-associated molecules in order to determine whether they can be expressed locally in significant amounts and whether the expression of genes coding for these molecules can change substantially as a function of age, damage, or ocular pathology. For example, quantitative PCR analyses determined the expression levels for three drusen-associated proteins: apolipoprotein E, vitronectin and C5, in the retina, EPR and choroid. The standardised mRNA rates for apolipoprotein E in the retina and EPR/choroid relative to the liver were 0.45 and 0.15, respectively, and the mRNA rates for vitronectin in the retina and EPR/choroid relative to the liver were 0.47 and 0.06. It should be noted, therefore, that the mRNA levels for both apolipoprotein E and vitronectin were almost 50% of those measured in the liver, and significantly higher than the rates in the brain compared to the liver (0.28 and 0.01). The liver-related C5 mRNA rates were 0.45±0.55 and 0.14±0.12 in the retina and EPR/choroid respectively.
ROLE OF MACROPHAGES IN DMLE
Ambati et al. describe retinal degeneration in mice that lack either the monocyte chemotactic protein 1 (MCP-1), a macrophage and memory T-cell chemotactic factor, or its ligand (CCR-2). Degeneration in these mice leads to the development of both neovascular (wet) and atrophic (dry) forms of AMD. These data support the hypothesis that inflammatory cells, in particular macrophages, may cause the damage in this disease.
The drusen of AMD are considered by some to be the residue of undigested material from malfunctioning phagocytic cells of the retinal epithelial layer. They appear to consist of 'waste' of various biological material: cholesterol-rich lipids, various proteins and more. In particular, oxidised lipoproteins have been identified within these structures, which would support the hypothesis that they could result from oxidative damage. Drusen also contain substances that are potentially chemotypic towards macrophages, such as components of the complement system and immunoglobulins.
Clinical studies show a correlation between drusen formation and autofluorescence (observed with a laser scanning ophthalmoscope), an indirect indication of the accumulation of granular deposits in the cellular layer of the EPR. These deposits, referred to as lipofuscin granules, are partially degraded cell membranes within lysosomes, and accumulate in cells that endocyte oxidised lipoproteins but cannot dispose of them adequately. This deficiency occurs both in scavenger cells predisposed to this lipoprotein disposal task, such as aged macrophages in atherosclerotic lesions (foam cells), and in less 'professional' phagocytes, such as neurons of the central nervous system and cells of the retinal epithelial layer. Defects in this function in retinal epithelial cells and choroid macrophages could contribute to the accumulation of drusen over time.
In addition to its already known proinflammatory activity, MCP-1 may also possess an immunomodulatory role. For instance, it appears to favour the shift of the T-cell immune response towards type II T-helpers by promoting the production of interleukin-4 (IL-4) and suppressing the cytokine IL-12 associated with the type I T-helper response. Blood-retinal barrier cells, including EPR cells, constitutively produce MCP-1 and can secrete large amounts of it after exposure to other cytokines and chemokines (such as IL-1B and tumour necrosis factor alpha).
The experience of Ambati et al. supports such a homeostatic and immunoregulatory role of MCP-1, as well as its chemotactic function towards proinflammatory or proangiogenic macrophages. MCP-1 could also coordinate the turnover of resident macrophages and immature dendritic cells, both of which form a cellular network in the choroid, which is intimately in contact with the epithelial cell layer. Immature dendritic cells mediate immunological tolerance and have been implicated in induced tolerance to antigens. Furthermore, MCP-1 appears to be required for the induction of immunological tolerance to antigens derived from the mucosal surface, which could be realised by induction of tolerance in dendritic cells.
It might make sense to consider that, in the eye, a decreased production of MCP-1, by senescent and dying cells of the retinal pigment epithelial layer, might affect the homeostatic and phagocytic function of resident macrophages and dendritic cells. Consequently, chemotactic agent-rich drusen could accumulate, with the ability to initiate a low-grade inflammatory response, leading to the recruitment of activated proangiogenic macrophages derived from the bone marrow. It would be these macrophages that could induce age-related macular degenerative disease.
GENETICS AND DMLE
The breakthrough in the history of inflammation occurred about a year ago, mainly due to human single-nucleotide polymorphisms (SNPs), subtle DNA sequence changes that can be used to detect disease-causing gene variants. In recent years, researchers have used SNPs to identify various chromosomal regions that contain genes that can influence the risk of obtaining AMD. In March 2006, three independent teams led by Josephine Hoh of Yale; Albert Edwards of Dallas; and Lindsay Farrer of Boston, reported what they had discovered about a gene on chromosome 1 that significantly increases the risk of getting AMD. The gene codes for a protein called Complement Factor H that keeps the complement system under tight control so that it does not attack healthy cells. The researchers found that people with a particular variant of the gene for Factor H were more likely to develop AMD. The high-risk variant could explain up to 50% of cases, presumably because the protein product of the altered gene is less effective in inhibiting the complement pathway. Genes related to age-related macular degeneration support the notion that inflammation plays a central role in damaging the central area of the retina in this vision disorder. Hageman et al. reported a link between a gene and the activation of the complement cascade. This gene, found on chromosome 6, produces a protein called factor B that is involved in complement activation.
Infection can be a determining factor in the development of AMD. Studies conducted on eyes of patients with exudative AMD, characterised by the development of blood vessels in the macula, showed patients with previous Chlamydia infection. A causal connection between Chlamydia infection and AMD needs to be confirmed, however.
Pericak-Vance et al. also propose the interaction between the LOC387715 gene and smoking as one of the most important environmental risk factors for AMD. The researchers found that the risk resulting from the association ¶ of smoking and carrying the gene variant was higher than the sum of the risk of the individual two factors in promoting the development of AMD.
Factor H and the B and LOC387715 genes are not the only genetic factors that are likely to influence the risk of developing AMD. For instance, in a study published in January, Pericak-Vance et al. relied on the analysis of eight genes that were found to be involved in the development of AMD. Their analysis also included VEGF. The VEGF gene product stimulates blood vessel development, which suggested that it may be involved in the formation of exudative AMD, which is the most severe form. But the complement genes and LOC387715 are certainly the main contributors to the risk of AMD. Based on this research, one can then seriously consider the use of preventive measures such as avoiding smoking, decreasing the intake of fatty foods and increasing the intake of antioxidants and carotenoids.
IDENTIFICATION OF FACTOR H
Hageman et al. claim that a variation in the gene for factor H (HF1/CFH) dramatically increases the likelihood of developing AMD, as well as membranous-proliferative glomerulonephritis type II (MPGN II). HF1 encodes for a protein involved in the body's first line of immunological defence (the innate system) against bacterial infections and other microbes. There are both protective and risk variants of this gene.
In earlier studies, Hageman, Mullins, Anderson and Johnson implicated the complement cascade, part of the innate immunological system, in the formation of drusen. Drusen contain EPR residues, dendritic cell processes and a variety of immune-associated molecules such as immunoglobulins, class II antigens, different complement components, activators and regulators. One of these regulators, factor H, is a key component of the alternative pathway of complement activation. All together these observations led the researchers to conclude that AMD, as well as other senile diseases such as Alzheimer's disease and atherosclerosis, may have a large inflammatory component involved.
The authors noted that MPGN II, if one leaves out its early onset, has ocular manifestations indistinguishable from AMD. A point mutation in the HF1 gene (I1166R) causes MPGN II in pigs and mice with severe factor H deficiency, which develop severe gromeluronephritis. Furthermore, affected individuals in two large families affected by MGPN III showed linkage to chromosome 1q31-32, a locus close to the 1q25-31 region that had previously been associated with AMD. All these observations led the researchers to consider factor H as the leading candidate for both AMDLE and MGPN II. The most frequent genetic variant at risk for developing AMD was associated with almost half of the 900 affected subjects in the study, while it appeared in 29% of the 400 controls (P=10-3). The strength of this association is far greater in comparison to genetic abnormalities previously attributed to AMD (ABCA4, FBNL5, FBNL6, APOE genes).
The first line of defence against microorganisms and other foreign particles is the complement system, which is responsible for recognising, attacking and killing invasive microorganisms by creating holes in their membranes. In some cases, however, sustained complement activation can lead to chronic inflammation, aggravate local tissue damage and contribute significantly to disease progression, as happens in Alzheimer's disease and atherosclerosis. To prevent such damage, certain proteins, including factor H, the main soluble inhibitor of the alternative pathway of complement activation, keep the system under tight control. Since most of the identified variations occupy important functional sites of the HF1 protein, the researchers suggested the possibility that these risk variants may alter the behaviour of the HF1 protein and hinder its role in regulating the complement pathway in the immune response.
It could therefore be argued that individuals with AMD and MPGN II share a functional defect in the factor H protein that affects the functionality of the complement system. Single nucleotide variations could, for example, change the binding of HF1 to the C3b fragment of complement or to C-reactive protein, sialic acid, or heparin. Similarly, these variations could alter the interaction between HF1 and microbes, possibly making tissues, such as EPR, more susceptible to infection. This concept is based on the fact that the most important activators of the alternative complement pathway are molecules that are readily found on many bacterial and viral surfaces. The final hypothesis is that a dysfunction of the complement system may result in local tissue damage, especially in the most vulnerable sites such as the renal glomerulus and macula. Hageman et al. also showed that the elastic layer of Bruch's membrane is preferentially thinner in the macular region. A disruption of this thin layer and subsequent inflammation could explain the predilection of the macula for lesion formation, including the formation of choroidal neovascular membranes.
The at-risk HF1 haplotype is associated with a wide phenotypic range of AMD, with the possible exception of geographic atrophy, observed in a small group of patients; it is therefore assumed that there is no distinct relationship between disease-associated genotype and clinical phenotype.
Furthermore, the results of this study are congruent with certain risk factors epidemiologically associated with AMD. For example, smoking inhibits factor H activity and increases the risk of AMD by 4 to 5 times.
The data of Hageman et al. also agree with previous statements on the role of complement in AMD. It seems likely that the inheritance of an HF1 haplotype at risk, in combination with an infectious agent or other atypical activators of the alternative pathway, such as immune complexes, amyloid beta peptide, or cholesterol, may substantially increase individual susceptibility to AMDLE and MPGN II.
It is clear that molecules involved in complement activation and its regulation will be future targets for the development of early diagnostic tests and therapeutic treatments for AMD and perhaps also for other diseases of inflammatory origin.
THE HTRA1 GENE
A locus on chromosome 10q26 has long been linked to the risk of AMD. A single nucleotide polymorphism, rs11200638, in the promoter region of the HTRA1 gene, is the gene variant most likely to be involved in the development of the disease, with an attributable risk of 49.3%. Immunohistochemical experiments unveiled, through the use of monoclonal antibodies, the presence of the HTRA1 product in the drusen of eyes with AMD. The single nucleotide polymorphism rs11200638 is located 512 bp (base pairs) upstream of the transcription start site of the HTRA1 gene. This gene encodes for a member of the serine protease family expressed in the retina and EPR (in mice). HTRA1 appears to regulate the degradation of proteoglycans in the extracellular matrix. This activity is thought to facilitate the access of matrix-degrading enzymes, such as collagenases and matrix matelloproteases, to their substrates. Overexpression of HTRA1 may alter the integrity of Bruch's membrane, promoting choroidal capillary invasion through the extracellular matrix, as occurs in exudative AMD. In addition, HTRA1 binds and inhibits transforming growth factor ? (TGF-?) an important regulator of extracellular matrix deposition and angiogenesis. DeWan et al. report the same association between this nucleotide variation of HTRA1 and exudative DMLE in the Chinese population.
TIMP-3 in Bruch's membrane: changes during ageing and in AMD
Matrix metalloproteases (MMPs) and their respective tissue inhibitors (TIMPs) play an important role in extracellular membrane (ECM) turnover. MMPs constitute a family of secretory enzymes, currently with more than 20 members, that are involved in degrading components of the ECM in the course of normal turnover and renewal. MMPs are also implicated in the early stages of neovascularisation, where it is thought that they, along with other proteases, may be required for the degradation of capillary membrane components as a prerequisite for new vessel formation. TIMPs, which are represented by four distinct gene products, are thought to suppress excessive ECM degradation and may play an important functional role in limiting neovascularisation. Timp-3, once secreted, binds to components of the ECM, while the other TIMPs do not.
One role of Timp-3 in Bruch's membrane may be as a potent local inhibitor of MMP activity, regulating Bruch's membrane turnover, as well as limiting choroidal neovascularisation.
Sorsby's fundus dystrophy is comparable to an early, hereditary onset of macular degeneration, characterised by thickening of Bruch's membrane and submacular neovascularisation. Mutations in the Timp-3 gene have been found in families with Sorsby's fundus dystrophy. Immunohistochemistry studies in a donated eye from a patient with Sorsby's showed an extensive accumulation of Timp-3 in the thickened Bruch's membrane. These observations led to an assessment of the presence of Timp-3 and its distribution in the thickened Bruch's membrane of eyes with AMD. Although no mutations in the coding region or in the regulatory elements of the Timp-3 gene have been discovered in patients with AMD observed so far, excess Timp-3 within the ECM could cause Bruch's membrane thickening and prevent normal membrane remodelling. Because of the importance of Bruch's membrane permeability in metabolite trafficking between choroid and EPR, it is important to understand the role of Timp-3 in normal ageing and AMD.
The study conducted by Motohiro Kamei and Joe G. Hollyfield analysed 36 normal eyes enucleated post mortem (between 14-96 years) and 15 eyes with AMD (between 74-98 years) and assessed the change in TIMP-3 distribution in relation to age. TIMP-3 levels in eyes of patients with AMD and healthy controls of the same age were also compared.
The study showed that immunoreactive Timp-3 was present in Bruch's membrane in every tissue sample from normal donors and was distributed throughout the entire thickness of the membrane. Although immunoreactivity was uniform in each donor sample, in general, the eyes of younger donors showed less immunoreactivity than the eyes of older donors. The immunoreactive Timp-3 was not evident in the neurosensory retina, choroid, or sclera.
Distribution and presence of TIMP-3 in eyes with AMD
Each eye with DMLE used contained soft drusen. Whenever soft drusen were observed, whether isolated or confluent, each was strongly immunoreactive with the anti-Timp-3 antibody. Hard drusen, when present, were also strongly immunoreactive.
In Bruch's membrane beneath the extensive areas of EPR atrophy, Timp-3 immunoreactivity was not evident or barely detectable. Choroidal neovascularisation was present in 8 of 15 eyes of donors with AMD. In areas where the EPR had proliferated around a choroidal neovascular membrane or fibroblastic scar, immunoreactive Timp-3 surrounded the hyperplastic EPR. The immunoreactivity was low and showed an irregular distribution, probably due to a loss of function and polarity of EPR proliferation.
In the transition regions between the areas of EPR atrophy and a normal EPR, there were decreases in Timp-3 immunoreactivity, but it was present in the soft drusen and under the central elastic layer of Bruch's membrane.
The amount of Timp-3 present in Bruch's membrane under the macula in the normal human retina appears to be age-dependent.
Sections from the fovea of donors from the second to third decade of life were weakly immunoreactive with the anti-TIMP-3 antibody. Furthermore, the distribution of Timp-3 changed in the ninth and tenth decades, with the immunoreactivity extending from Bruch's membrane to the choriocapillaris.
Quantitative analysis showed that the content and function of Timp-3 in the macula increased with age with a significant correlation coefficient (r = 0.66 and 0.67, respectively). Timp-3 is therefore an age-related protein. Quantitative analysis indicated that Timp-3 levels were significantly elevated in the macula of eyes with AMD compared to normal eyes.
Eyes with DMLE, however, showed a non-uniform distribution, with a virtual absence of Timp-3 immunoreactivity in areas of EPR atrophy and abundant immunoreactivity in areas outside the atrophic areas where EPR was present. This indicates that the distribution of Timp-3 in AMD in eyes with EPR atrophy is not uniform.
Various hereditary or non-hereditary factors such as protein alterations, oxidative stresses, or hydrolytic enzyme disorders can contribute to the accelerated accumulation of these residues with age. Since Timp-3 largely inhibits MMPs, drusen with an excess of Timp-3 may delay Bruch's membrane renewal. This may result in Bruch's membrane thickening, reduced permeability of Bruch's membrane to metabolite and nutrient trafficking between the choroid and the EPR, and subsequent atrophy of the EPR and photoreceptors.
In areas where choroidal neovascularisation was observed, EPR was absent and no immunoreactivity for Timp-3 was evident in the underlying Bruch's membrane.
We conclude that Timp-3 content in Bruch's membrane in macula increases during normal ageing and that Timp-3 content is elevated above normal levels in the macular region of eyes with AMD. This suggests that Timp-3 may be one of the key molecules in Bruch's membrane thickening in normal ageing and AMD.
Systemic indicators of inflammation, endothelial dysfunction and AMD
It has been hypothesised that inflammation plays a role in the pathogenesis of AMD. Hageman et al. indicated that drusen contain proteins related to immune-mediated processes and inflammation. Chronic inflammatory cells have been observed on the outer surface of Bruch's membrane in eyes with age-related macular degeneration of the exudative type. These cells can cause atherogenic and microvascular lesions by direct release of oxidants, toxic oxygen compounds and proteolytic enzymes that can also damage Bruch's membrane. However, data from epidemiological studies observed so far have not indicated a consistent relationship between systemic inflammation or the presence of inflammatory markers and AMD.
Endothelial dysfunction, derived from inflammation, hypertension, smoking and other factors, has been considered a prerequisite in the pathogenesis of AMD. The intent of Klein et al.'s study was to investigate whether markers of systemic inflammation and endothelial dysfunction were associated with AMD. The procedures followed in this study included: weight, height and systemic blood pressure, a standardised questionnaire, which included specific questions regarding a history of emphysema, arthritis, smoking, alcohol consumption and vitamin use.No evidence was found for the association of these markers of systemic inflammation with AMD, in agreement with the data from the cardiovascular study (CHS). This lack of association between the studied indicators of systemic inflammation (serum hsCRP, SAA, Tnf-? and IL-6) and AMD concurs with the demonstrated ineffectiveness of the use of systemic anti-inflammatory drugs. Clinical trials have instead suggested the possible beneficial effects of intravitreally administered corticosteroids. Intravitreal corticosteroids reduce the incidence of laser-induced neovascular membranes in primates by reducing the activity of inflammatory cells and their number in the choroid. Other potential mechanisms include reduction of VEGF expression and down-regulation of intracellular adhesion molecule 1 (ICAM-1) expressed on EPR cells and vessel endothelium, which regulates leukocyte adhesion and diapedesis during inflammation.
CONCLUSIONS
Five summary concepts relevant to the pathology of AMD
Firstly, age-related changes with the addition of pathological events lead to AMD.
Secondly, oxidative stress over the years and in AMD causes damage to the EPR and choriocapillaris.
Third, EPR and choriocapillary damage in AMD results from chronic inflammation of the choroid and Bruch's membrane.
Fourth, damage to the EPR, choriocapillaris and inflammation can lead to abnormalities of the extracellular matrix (ECM). This abnormal ECM causes impaired diffusion of nutrients from the EPR to the neuroretina, which can lead to further retinal and EPR damage.
Fifth, abnormalities in the extracellular matrix (ECM) lead to an alteration in the function of the EPR-coriocapillary complex that evolves towards retinal atrophy, EPR atrophy and new vessel growth (CNV).
In this sequence of events, environment and genetics can alter the course of the disease.
Dr Laura Bertazzi
Dr. Cristiano Luiselli
Dr Pierfilippo Sabella
Chair of Clinical Ophthalmology
Department of Clinical Sciences 'Luigi Sacco' - Milan
Ophthalmology Operating Unit 'Luigi Sacco' Hospital - Milan
BIBLIOGRAPHY
1. Klein R et al. Prevalence of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology 99: 933-943, 1992.
2. Kahn HA et al. The Framingham Eye Study Outline and major prevalence findings. Am J Epidemiol 106: 17-32, 1977.
3. Seddon JM, George S, Rosner B, Rifai N. Progression of age-related macular degeneration: prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch Ophthalmol. 2005 Jun;123(6):774-82.
4. Donoso LA, Kim D, Frost A, Callahan A, Hageman G. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006 Mar-Apr;51(2):137-52.
5. Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004 Apr;122(4):598-614.
6. Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in ageing and age-related macular degeneration. Prog Retin Eye Res. 2001 Nov;20(6):705-32
7. Penfold PL, Killingsworth MC, Sarks SH. Senile macular degeneration: the involvement of immunocompetent cells. Graefes Arch Clin Exp Ophthalmol. 1985;223(2):69-76.
8. Forrester JV. Macrophages eyed in macular degeneration. Nat Med 2003 Nov;9(11):1350-1
9. Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003 Nov;9(11):1390-7.
10. Wiggs JL. Complement factor H and macular degeneration: the genome yields an important clue. Arch Ophtalmol. 2006 Apr;124(4):577-8.
11. Despriet DD, Klaver CC, Witteman JC, Bergen AA, Kardys I, de Maat MP, Boekhoorn SS, Vingerling JR, Hofman A, Oostra BA, Uitterlinden AG, Stijnen T, van Duijn CM, de Jong PT. Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. JAMA. 2006 Jul 19;296(3):301-9.
12. Haines JL, Schnetz-Boutaud N, Schmidt S, Scott WK, Agarwal A, Postel EA, Olson L, Kenealy SJ, Hauser M, Gilbert JR, Pericak-Vance MA. Functional candidate genes in age-related macular degeneration: significant association with VEGF, VLDLR, and LRP6. Invest Ophthalmol Vis Sci. 2006 Jan;47(1):329-35.
13. Emil Wirostko, William J. Wirostko, Barbara M. Wirostko. Age-related macular degeneration is an inflammatory disease possibly treatable with minocyclineActa Ophthalmol Scand. 2004 Apr;82(2):243-4.
14. Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, Cameron DJ, Chen H, Zhao Y, Pearson E, Li X, Chien J, Dewan A, Harmon J, Bernstein PS, Shridhar V, Zabriskie NA, Hoh J, Howes K, Zhang K. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006 Nov 10;314(5801):992-3.
15. Kamei M, Hollyfield JG. TIMP-3 in Bruch's membrane: changes during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1999 Sep;40(10):2367-75.
16. Bailey TA, Alexander RA, Dubovy SR, Luthert PJ, Chong NH. Measurement of TIMP-3 expression and Bruch's membrane thickness in human macula. Exp Eye Res. 2001 Dec;73(6):851-8.
17. Strunnikova N, Hilmer S, Flippin J, Robinson M, Hoffman E, Csaky KG. Differences in gene expression profiles in dermal fibroblasts from control and patients with age-related macular degeneration elicited by oxidative injury. Free Radic Biol Med. 2005 Sep 15;39(6):781-96
18. Nakata K, Crabb JW, Hollyfield JG. Crystallin distribution in Bruch's membrane-choroid complex from AMD and age-matched donor eyes. Exp Eye Res. 2005 Jun;80(6):821-6.
19. Bok D. Evidence for an inflammatory process in age-related macular degeneration gains new support. Proc Natl Acad Sci U S A. 2005 May 17;102(20):7053-4.
20. Penfold PL, Killingsworth MC, Sarks SH. Senile macular degeneration. The involvement of giant cells in atrophy of the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1986 Mar;27(3):364-71.
21. Penfold P, Killingsworth M, Sarks S. An ultrastructural study of the role of leucocytes and fibroblasts in the breakdown of Bruch's membrane. Aust J Ophthalmol. 1984 Feb;12(1):23-31.
22. Vine AK, Stader J, Branham K, Musch DC, Swaroop A.Biomarkers of cardiovascular disease as risk factors for age-related macular degeneration. Ophthalmology. 2005 Dec;112(12):2076-80.
Dr. Carmelo Chines
Direttore responsabile