



Difficult to diagnose, but sensitive to early treatment
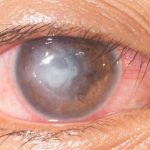
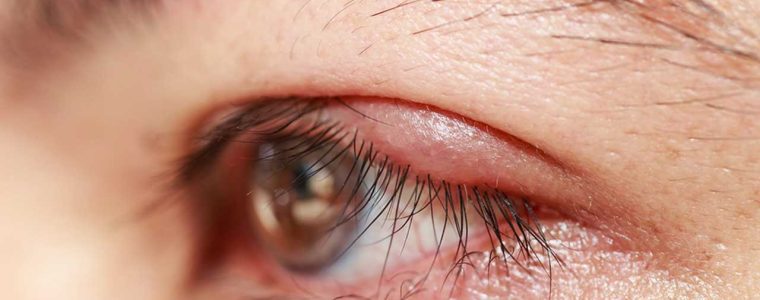
Eyes that often become inflamed, in young patients, with many of the clinical signs of keratitis, but with a peculiarity: inflammation of the ocular surface is associated with a significant reduction in hearing ability. The diagnostic hypothesis to consider is: Cogan's syndrome, a rare disease with an autoimmune aetiology, so called because it was first identified in 1945 by David G. Cogan who described the cases of 4 patients as "non-syphilitic interstitial keratitis accompanied by vestibulo-auditory symptoms"..
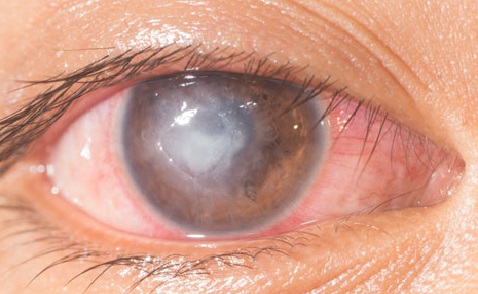 The disease predominantly affects young adults, with an age of onset between 20 and 30 years, while it is uncommon in children. The clinical onset is characterised systemically by fever, fatigue and weight loss. Frequent occurrences are auditory symptomssuch as tinnitus and reduced hearing, and ocular symptomssuch as photophobia, oscillopsia and recurrent inflammation of the cornea.
The disease predominantly affects young adults, with an age of onset between 20 and 30 years, while it is uncommon in children. The clinical onset is characterised systemically by fever, fatigue and weight loss. Frequent occurrences are auditory symptomssuch as tinnitus and reduced hearing, and ocular symptomssuch as photophobia, oscillopsia and recurrent inflammation of the cornea.
Interstitial keratitis often manifests itself with a ring-shaped infiltrate, which may therefore spare the pupil area and for this very reason the first symptom of Cogan's syndrome may not be a visual disturbance.
At the onset, hearing loss may occur first in one ear and then in the other, and later become bilateral. It may be associated with balance disorders, which often lead to the diagnosis of Ménière's Disease.
The ocular, auditory and/or balance manifestations in the early stages of the disease may not be concomitant and it is precisely this that makes diagnosis very difficult if the specialist is not familiar with the clinical picture of Cogan's Syndrome.
In 1981, Haynes et al. also proposed to distinguish two forms:
– Typical' Cogan syndrome, characterised by sensorineural hearing loss associated with interstitial keratitis
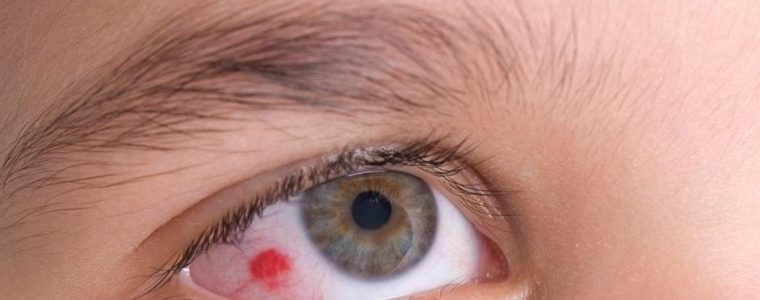
– Atypical' Cogan syndrome, characterised by sensorineural hearing loss associated with inflammation of any part of the eyeball (scleritis, episcleritis, uveitis). In the 'atypical' form, interstitial keratitis may be present, but not necessarily.
The aetiology is autoimmune and the inflammation in the eye and ear is due to the patient's immune system, which produces autoantibodies that attack the inner ear and ocular tissue.
Diagnosis
There are no specific laboratory tests to diagnose Cogan syndrome. Anti-Cogan antibody titration, which has a sensitivity of 50%, may be useful, so that the negativity of the test does not allow the disease to be ruled out, while its positivity may support the diagnosis.
Recent studies suggest MRI to rule out neurinoma of the auditory nerve. However, MRI may not provide relevant results.
Diagnosis is essentially clinical, so the medical history plays a crucial role and a thorough and thorough slit-lamp examination can be decisive.
Therapy
Corticosteroids are the gold standard for the treatment of Cogan's syndrome. Topical glucocorticoids, in combination with cycloplegics, can be used for the treatment of mild, isolated ocular pathology while, when ocular involvement is more severe, hearing is impaired and systemic symptoms are present, systemic corticosteroids are recommended. Usually, systemic high-dose corticosteroid therapy (daily administration of 1-1.5 mg/kg prednisone) prevents deafness and shows benefits within 2-3 weeks. However, the short-term benefits of steroid therapy are associated with the risk of severe side effects. Consequently, in patients with severe and refractory disease, it is recommended to consider second-line treatment with immunosuppressants. Conventional immunosuppressants, such as methotrexate, cyclophosphamide, azatiotropin or cyclosporin A, have low efficacy, while the number of documented cases of positive response to infliximab, a tumour necrosis factor alpha (TNFalpha) inhibitor, is increasing. Patients treated with infliximab showed an improvement in cochleo-vestibular symptoms that allowed a gradual reduction in corticosteroid dosage, with a significant difference compared to patients treated only with steroids or DMARDs. The early use of infliximab, as first-line treatment in the most severe cases, seems to have even greater efficacy. Cochlear implants represent a useful surgical option in patients with severe sensorineural deafness who do not respond to intensive treatment with immunosuppressants.
The prognosis depends mainly on the risk of permanent deafness and cardiovascular complications, particularly aortic insufficiency. Severe involvement of internal organs and deaths related to cardiovascular complications are, however, rare.
Conclusion
Cogan's Syndrome is a curable diseasewhich responds well to early immunosuppressive treatment. It should, therefore, be known very well by the ophthalmologist, ENT specialist, internist and paediatrician. Unfortunately, however, to this day Cogan's Syndrome remains a rare disease that is often misunderstood, because only a few ophthalmologists and ENT specialists correlate ocular inflammation and neuro-sensory deafness.
Gluth Mb, Baratz Kh, Matteson El, Driscoll Cl, Cogan Syndrome: A Retrospective Review of 60 Patients Throughout a Half Century, in Mayo Clinic proceedings, 2006 Apr.
Haynes Bf, Kaiser-Kupfer Mi, Mason P, Fauci As, Cogan Syndrome: Studies in Thirteen Patients, Long-Term Follow-Up, and a Review of the Literature, in Medicine, 1980 Nov. URL
Iliescu DA, Timaru CM, Batras M, De Simone A, Stefan C. COGAN'S SYNDROME. Rom J Ophthalmol. 2015 Jan-Mar;59(1):6-13. PMID: 27373108; PMCID: PMC5729811.