






Retinoblastoma is a rare form of tumour, which affects the retina and typically arises in children before the age of five.
Globally, it affects about 1 in every 15,000 children and it is estimated that between 8,600 and 9,000 are newly diagnosed each year. Unfortunately, about 90% of children with this neoplasm live in developing countries.
Retinoblastoma is a disease of genetic origin related to mutations in the Rb1 gene, located on chromosome 13. The retinoblastoma protein (pRb or Rb), in fact, is a tumour suppressor protein identified in 1991, which has been found to be non-functional in several types of cancer. Rb's normal function is to block the cell at one stage of the cell cycle, preventing it from dividing incorrectly or damagingly.
Retinoblastoma develops when both alleles of the RB1 gene, which encodes the Rb protein, are inactivated by a mutation.
Retinoblastoma can be transmitted by the parents in familial hereditary forms (autosomal dominant transmission) or can arise 'ex novo' during embryogenesis in non-familial hereditary forms.
Diagnosis
The diagnosis of retinoblastoma is clinical, through certain typical signs, in particular the whitish reflex of the pupils of the eyes affected by the neoplasm. It is often discovered when a flash photo is taken of the child and the 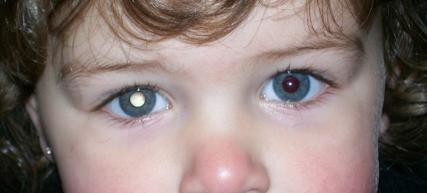 pupils appear white instead of the normal reddish-orange reflection. This sign is called "leucocoria" or 'cat's eye pupil'.
pupils appear white instead of the normal reddish-orange reflection. This sign is called "leucocoria" or 'cat's eye pupil'.
It can also induce squinting or symptoms such as redness and soreness in the eyes. The latter form of pseudo-inflammation is found in about one-tenth of patients, while less common symptoms are a protruding eyeball, discolouration of the iris of the affected eye, the presence of differences in pupil size, shape and activity, and other metastasis-related symptoms such as bone pain.
Visual acuity may be impaired and the child may complain of double vision.
The diagnosis must be confirmed with a indirect ophthalmoscopyto be performed in mydriasis and under anaesthesia in the case of very young children or infants. The entire inner eye cavity, the entire retina and the optic nerve head must be carefully examined. A slit-lamp biomicroscopyusing a powerful light beam and a microscope. Further investigations may include fluorangiography, ultrasonography of the eyeball, and magnetic resonance imaging to obtain a detailed image of the soft tissue in order to stage the spread of the tumour.
OCT, optical computed tomography, could be useful, but it is preferred to avoid it in very young children so as not to expose them to the risk of exposure to ionising radiation, especially if they have a family history of retinoblastoma and other tumours.
Therapy
The therapeutic approach to retinoblastoma depends on a variety of factors, specifically:
1. tumour size
2. number of tumour sites
3. location of the tumour
4. extension and involvement of intraocular and extraocular structures
5. presence of other secondary tumours, since lung or bladder tumours, sarcomas or osteosarcomas and melanomas are fairly frequent in individuals with hereditary forms of retinoblastoma.
6. family history of retinoblastoma
7. visual residue preserved
This ocular neoplasm must be managed by a healthcare team highly specialised in the management of paediatric tumours. Diagnosis must be as early as possible and the primary aim of treatment must be to prevent death from the tumour, save the child's eye and, as far as possible, preserve visual function, with minimal side effects.
Currently used therapeutic approaches include:
1. Cryotherapy: A cryoprobe is used to freeze and destroy tumours up to 5 mm in diameter and 3 mm thick. It may be necessary to repeat this treatment at 3-4 week intervals.
2. Laser photocoagulationlaser light energy is used to superheat the tumour and destroy the vascular network that feeds it, rendering it inert or in some cases attackable with other ablative techniques.
3. Thermotherapy: microwaves are used to heat cancer cells and destroy them.
4. Radiotherapy: Both external irradiation and brachytherapy are used. Irradiation has the advantage of greater precision, which spares the surrounding tissue and thus better preserves vision. Three modalities are currently used: IMRT (Intensity Modulated Radiation Therapy), Gamma Knife and proton beam therapy. Brachytherapy involves the use of plaques, on which radioactive elements are placed, which are sutured to the sclera at the tumour. The plaque is left in place for 3 to 7 days depending on the size and extent of the tumour. Radiotherapy carries serious risks of side effects on the brain and eye.
5. Chemotherapy: Systemic chemotherapy is used when distant metastases have occurred. The drugs used include cisplatin, etoposide and vincristine. It may be administered intravenously, orally, arterially (through the artery feeding the eyeball - only for tumours confined to the eye) or intracranially (into the space surrounding the central nervous system and containing cerebrospinal fluid).
6. Enucleation: surgical procedure involving the removal of the eye and part of the optic nerve. It is only used for larger and more extensive tumours, often as a second-line treatment if irradiation or other therapies aimed at saving the eye have failed, and only when there is no hope of saving sight. It is often associated with the implantation of an artificial eye the size of an adult eye and which is reattached to the eye muscles to ensure more natural movements. Children in this case must be monitored for at least two years to detect any recurrence.
7. Exenteratio Orbitae: removal of the contents of the orbit including the ocular adnexa, i.e. the eyeball, extrinsic musculature, peribulbar fat, periorbita, eyelids and possibly the periocular skin. It represents the most invasive and destructive procedure.
Survival
Incidence and survival rates vary substantially from country to country and are related to the earliness of diagnosis. An epidemiological study just published in Jama Ophthalmology, EUROCARE-6 Working Group. Survival and health care burden of children with retinoblastoma in Europe analysed data on children diagnosed with retinoblastoma, collected from 81 cancer registries in 31 European countries participating in the EUROCARE-6 project, covering the 14 years between January 2000 and December 2013.
The incidence of retinoblastoma in Europe has remained stable at 4 cases per million children, aged 0-14 years, with a life expectancy at 5 years of 97.8%
Worldwide, on average, 9 out of 10 children diagnosed with retinoblastoma can be cured and do not have tumour recurrences within 5 years after treatment.
Lifelong follow-up is necessary because of the high risk of secondary tumours or of developing a tumour in the contralateral eye. The latter usually arises within the first 3 years after the first tumour appears, so the eyes must be examined every 2-4 months for at least the first 28 months.
On the subject of paediatric ophthalmology, see also
- COVID-19: long-term effects in children - Oculista Italiano
- The 'digital health' of children and young people - Oculista Italiano
- Retinal trauma in children: watch out for the ball - Oculista Italiano
- Convergence insufficiency in children: how to intervene - Oculista Italiano
- Chévez-Barrios P, Chantada GL, Wilson MW. Incidence and Survival Rates in European Children With Retinoblastoma. JAMA Ophthalmol. 2024;142(11):1071–1072. doi:10.1001/jamaophthalmol.2024.4268
- Virgili G, Capocaccia R, Botta L, et al; EUROCARE-6 Working Group. Survival and health care burden of children with retinoblastoma in Europe. JAMA Ophthalmol. Published online October 10, 2024. doi:10.1001/jamaophthalmol.2024.4140