


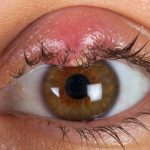
L’orzaiolo è una condizione oculare molto comune e frequente, che può causare fastidio e preoccupazione, ma con le giuste informazioni è possibile gestirlo efficacemente. Questo disturbo si manifesta come un piccolo rigonfiamento doloroso sulla palpebra, spesso causato da un’infezione batterica o dall’ostruzione delle ghiandole sebacee. Comprendere le cause principali e i fattori di rischio associati può aiutare a prevenirne l’insorgenza e a ridurre il disagio. In questa guida, esamineremo le origini dell’orzaiolo, offrendo consigli pratici per evitarlo e per trattarlo in modo efficace quando si presenta. Scopriamo insieme come mantenere i nostri occhi sani e liberi da fastidi.
Cos’è l’Orzaiolo
L’orzaiolo è una infiammazione benigna delle palpebre, che rappresenta una delle condizioni oculari più comuni.
È importante capire di cosa si tratta per affrontarlo nel modo migliore. In questa sezione, esploreremo l’eziologia dell’orzaiolo, i sintomi caratteristici e le diverse tipologie che si possono manifestare.
Definizione e Sintomi
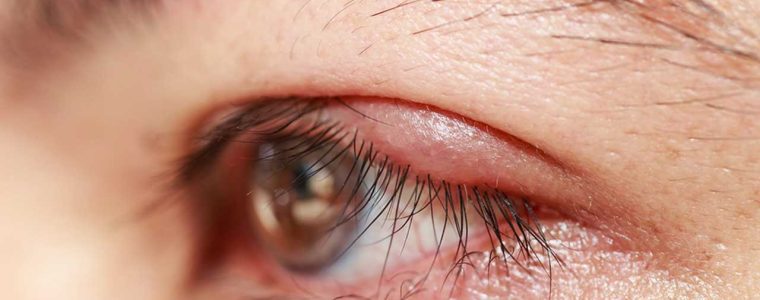
L’orzaiolo si presenta come un piccolo rigonfiamento doloroso sulla palpebra, spesso causato da un’infezione batterica.
Questo gonfiore è dovuto all’infiammazione delle ghiandole sebacee o sudoripare situate vicino alla base delle ciglia.
Tra i sintomi più comuni che si possono manifestare, ricordiamo:
- Dolore e arrossamento nella zona interessata
- Sensazione di corpo estraneo nell’occhio
- Lacrimazione eccessiva
Il rigonfiamento è solitamente ben delimitato e può contenere pus.
È di fondamentale importanza riconoscere questi sintomi per intervenire tempestivamente.
Tipologie di Orzaiolo
Esistono principalmente due tipi di orzaiolo: esterno e interno.
Quello esterno si sviluppa sulla parte esterna della palpebra, mentre l’interno si forma all’interno della palpebra stessa.
- Orzaiolo Esterno: Colpisce le ghiandole sebacee alla base delle ciglia. È più visibile e spesso meno
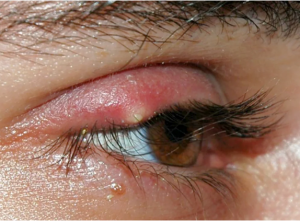
Orzaiolo esterno doloroso.
- Orzaiolo Interno: Coinvolge le ghiandole di Meibomio, risultando più doloroso e meno evidente.
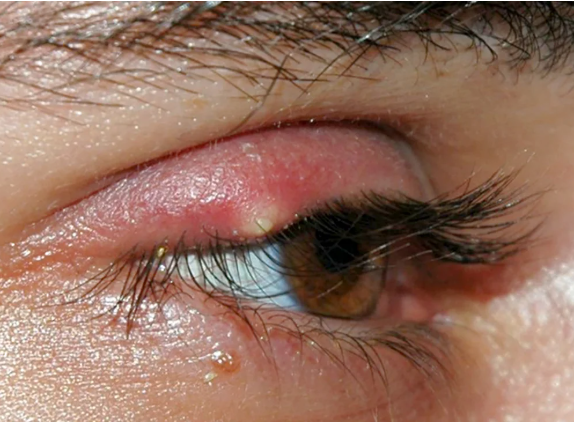
Comprendere la differenza tra queste tipologie aiuta a determinare il trattamento più adeguato.
Cause Principali dell’Orzaiolo
Identificare le cause dell’orzaiolo è essenziale per prevenirne l’insorgenza. Analizzeremo sia i fattori ambientali che le predisposizioni genetiche che possono contribuire a questa condizione.
Fattori Ambientali e Abitudini
L’ambiente e le abitudini personali giocano un ruolo cruciale nello sviluppo dell’orzaiolo.
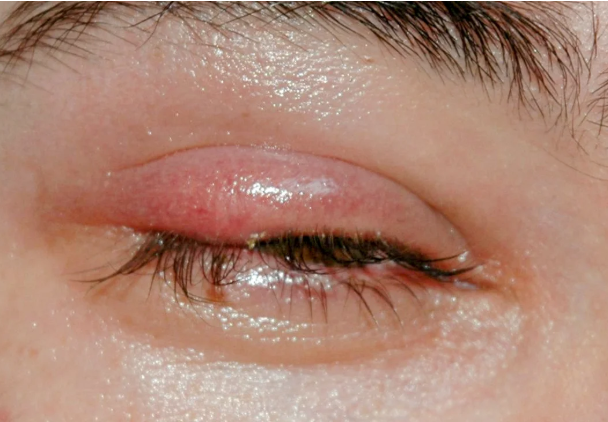
L’esposizione a polvere, vento e inquinamento può aumentare il rischio di infezioni oculari.
Anche l’uso di cosmetici scaduti o non adeguatamente rimossi può contribuire.
- Mani sporche: Toccare gli occhi senza lavarsi prima le mani può trasferire batteri nell’area perioculare e favorire l’insorgenza dell’orzaiolo.
- Uso di lenti a contatto: Lenti non adeguatamente pulite, mal conservate o scadute possono causare irritazioni sia alla superficie dell’occhio che in area perioculare..
Adottare buone pratiche igieniche è fondamentale per la prevenzione.
Predisposizioni
Alcuni individui sono più predisposti a sviluppare orzaioli, a causa della loro corredo genetico. Infatti, se in famiglia ci sono precedenti frequenti di orzaiolo, il rischio aumenta.
Anche condizioni mediche come la dermatite seborroica e l’acne rosacea possono influenzare la frequenza degli orzaioli.
Quindi possiamo riepilogare tra tra le condizioni predisponenti:
- Ereditarietà: Un fattore importante da considerare.
- Condizioni dermatologiche preesistenti: Possono aumentare la suscettibilità alle infezioni.
Essere consapevoli di queste predisposizioni consente di prendere misure preventive più mirate.
Riconoscere i Segnali
I sintomi dell’orzaiolo possono variare in intensità, ma alcuni segnali sono comuni.
Tipicamente, il primo sintomo è un piccolo gonfiore rosso e doloroso sulla palpebra.
Altri sintomi includono:
- Sensazione di prurito o bruciore.
- Lacrimazione eccessiva.
- Sensibilità alla luce.
Riconoscere questi sintomi precocemente permette di adottare misure per alleviare il disagio e accelerare la guarigione.
Quando Preoccuparsi
Mentre gli orzaioli sono generalmente innocui, ci sono situazioni in cui è necessario preoccuparsi.
Un orzaiolo che non migliora entro una settimana può indicare un’infezione più grave.
Segnali d’allarme includono:
- Aumento del dolore e del gonfiore.
- Febbre o brividi.
- Visione offuscata.
In questi casi, è essenziale consultare tempestivamente un medico oculista per valutare il trattamento appropriato.
Metodi di Prevenzione
Prevenire l’orzaiolo è possibile con alcune semplici misure. Esamineremo l’importanza dell’igiene personale e come uno stile di vita sano possa contribuire alla salute oculare.
Igiene Personale e Cura
Una corretta igiene personale è la prima linea di difesa contro l’orzaiolo.
- Lavarsi le mani frequentemente.
- Evitare di toccare gli occhi con le mani sporche.
- Rimuovere accuratamente il trucco prima di dormire.
Queste semplici azioni riducono significativamente il rischio di infezioni.
Alimentazione e Stile di Vita
Uno stile di vita sano supporta la salute degli occhi.
Una dieta bilanciata ricca di vitamine può rafforzare il sistema immunitario e ridurre l’incidenza di infezioni oculari.
- Vitamina A: La vitamina A, o retinolo, è una vitamina liposolubile essenziale per la vista, la crescita e il normale sviluppo dei tessuti, oltre a contribuire al funzionamento del sistema immunitario. Si trova principalmente in alimenti di origine animale, ma anche nel latte e nei suoi derivati (burro e formaggio) e nelle uova. In molti alimenti di origine vegetale sono contenuti, invece, i carotenoidi, precursori della vitamina A. I carotenoidi sono presenti in particolare in frutta e verdura di colore rosso, giallo e arancione, come albicocche, carote, anguria, frutti di bosco e pomodori.
- Omega-3: Gli Omega-3 sono acidi grassi polinsaturi essenziali, cioè devono essere assunti con l’alimentazione perché il corpo non li produce. Sono importanti per la salute del cuore, del cervello e in generale per la prevenzione di molte patologie. Gli omega-3 sono contenuti principalmente nel pesce, nella frutta secca, come noci e nocciole, ma soprattutto in alcuni oli (principalmente in olio di lino e di semi di lino)
Integrare questi nutrienti nella dieta quotidiana può fare la differenza nella prevenzione.
Rimedi Naturali e Trattamenti
Quando si presenta un orzaiolo, è importante sapere come trattarlo efficacemente. Questa sezione esplora i rimedi che è possibile adottare in ambiente domestico e esamina le situazioni in cui è necessario consultare un medico.
Trattamenti Domestici
I trattamenti domestici possono essere efficaci nei casi lievi di orzaiolo.
Vengono spesso raccomandati impacchi caldi, da praticare secondo queste modalità:
- Applicare un panno caldo sull’occhio chiuso per 10-15 minuti, tre volte al giorno.
- Massaggiare delicatamente la palpebra per favorire il drenaggio.
Questi semplici rimedi possono alleviare il dolore e accelerare la guarigione.
Quando Consultare un Medico
Sebbene molti orzaioli possano essere trattati a casa e si risolvono spontaneamente, ci sono situazioni in cui è fondamentale rivolgersi a un professionista.
Se i sintomi persistono per più di una settimana o se si verificano complicazioni, è consigliabile consultare un medico.
In particolare, in caso di dolore intenso o peggioramento dei sintomi, è fondamentale cercare assistenza medica.
Un medico può determinare se sono necessari trattamenti più specifici, quali:
- Antibiotici: In casi di infezioni persistenti o severe.
- Incisione e drenaggio: In casi rari, il piccolo intervento chirurgico deve essere eseguito da uno specialista.
Il trattamento successivo varia a seconda dei sintomi e della risposta al trattamento iniziale.
Miti e Verità sull’Orzaiolo
Esistono molti miti riguardo all’orzaiolo che possono confondere. In questa sezione, sfateremo alcuni dei falsi miti più comuni e forniremo verità scientifiche affidabili.
Falsi Miti Comuni
Molte persone credono che l’orzaiolo sia contagioso, ma non è così.
Altri pensano che spremere un orzaiolo possa aiutare, ma in realtà può peggiorare l’infezione.
- Non è contagioso: L’orzaiolo deriva da infezioni batteriche locali.
- Non spremere: Può causare ulteriori infezioni.
Conoscere la verità aiuta a gestire meglio la situazione.
Verità Scientifiche e Consigli Affidabili
Le verità scientifiche sull’orzaiolo possono guidare le azioni preventive e i trattamenti. È importante seguire consigli medici basati su prove:
- Igiene è fondamentale: Lavarsi le mani è la miglior prevenzione.
- Consultare un medico: Essenziale se i sintomi persistono.
Seguendo questi consigli, si può ridurre l’incidenza e la gravità degli orzaioli.
In materia di infezioni degli annessi oculari vedi anche:
- Calazio: cause e rimedi – Oculista Italiano
- La dermatite palpebrale: come prevenirla – Oculista Italiano
- Manifestazioni oculari delle malattie dermatologiche: disturbi infettivi e infiammatori – Oculista Italiano
- Willmann D, Guier CP, Patel BC, Melanson SW. Hordeolum (Stye). 2024 Dec 11. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 29083787.
- Carlisle RT, Digiovanni J. Differential Diagnosis of the Swollen Red Eyelid. Am Fam Physician. 2015 Jul 15;92(2):106-12. PMID: 26176369.